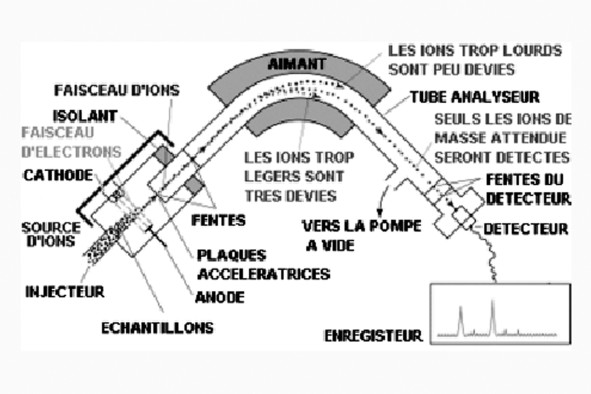
Article paru dans le Bulletin du G.E.S.T., N° 172, mars 2012
Robert Six
I. RADIOACTIVITE DES ROCHES SEDIMENTAIRES
A. Origine des roches sédimentaires
La plupart des roches sédimentaires se forment en milieu aqueux (lacs, mers, océans) et sont composées principalement d’éléments clastiques ou détritiques, qui se sont sédimentés. Ces divers constituants proviennent de la désagrégation, par différents procédés d’érosion, de roches préexistantes : roches magmatiques, métamorphiques et même sédimentaires.
L’érosion sera causée par des phénomènes physiques et mécaniques ou des procédés chimiques ou l’action d’organismes vivants.
On distingue traditionnellement trois origines différentes de sédiments : détritiques, chimiques et biologiques. La distinction ne repose pas sur l’origine première des matériaux mais sur les modalités de transport et de dépôt.
Les éléments détritiques arrachés aux roches du sol sont entraînés par les eaux de ruissellement, l’avancée des glaciers ou les vents. Durant leur transport, ils subiront une usure plus ou moins prononcée selon leur dureté.
Les procédés chimiques peuvent intervenir en présence d’eau lorsque celle-ci est plus ou moins chargée de sels de natures diverses.
Les actions organiques sont souvent causées par des bactéries ou des végétaux inférieurs sous forme d’action chimique, ou par des organismes supérieurs, dont l’action est surtout mécanique.
B. Uranium et thorium dans les roches sédimentaires
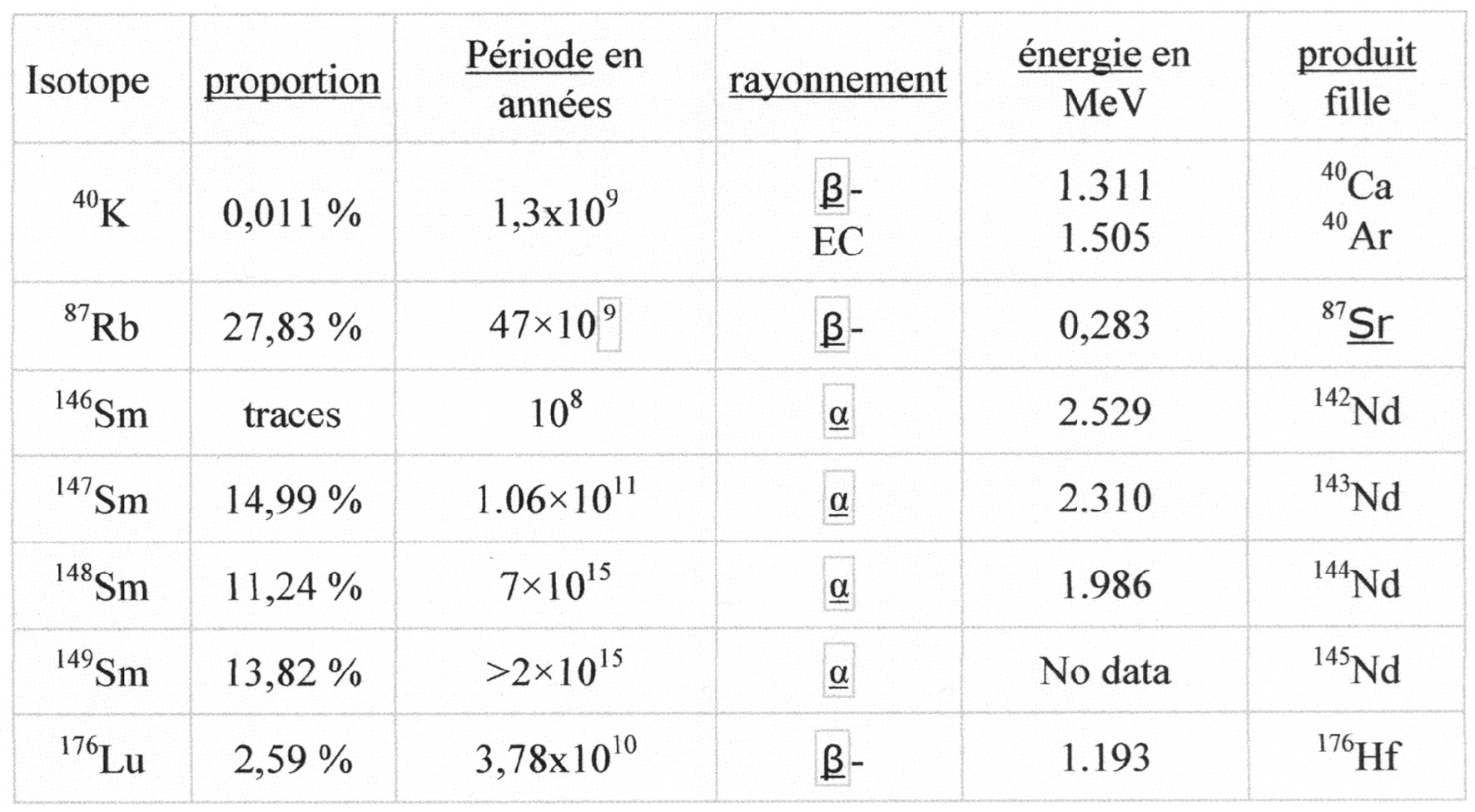
Rappelons que la radioactivité naturelle des sols et des roches est due à la présence des trois radioéléments naturels à longue durée de vie : 238U, 232Th, 40K et de leurs produits dérivés comme le radium. Selon leur nature, les sols et les roches ont une radioactivité spécifique qui permet de les différencier. Ainsi, les argiles et certains sables résultant de la désagrégation des massifs primaires sont plus radioactifs que les craies et les calcaires.
L’uranium et le thorium des roches sédimentaires proviennent donc directement ou indirectement des roches magmatiques. Du fait de leur densité élevée (U = 18,95 ; Th = 11,72), ils se déposeront partiellement au pied de la roche-mère, ou pendant le transport, ou atteindront le bassin de sédimentation. Les éléments radioactifs, étant de ce fait fortement dispersés, les roches sédimentaires en contiendront moins que la roche-mère. Une roche sédimentaire, déjà pauvre en U et Th, désagrégée à son tour par l’érosion, donnera des sédiments encore moins riches en ces éléments. Il en résulte que l’U et le Th des roches sédimentaires est, en général, à par quelques placers, à l’état diffus, et que l’on ne trouve pas de gisements uranifères ou thorifères à forte concentration dans ce type de roches.
Les minéraux microscopiques uranifères et thorifères résistent mal à l’érosion. Parmi les plus fragiles citons l’autunite, la pechblende, la samarskite, la thorianite et la thorite. Ils seront détruits avant la sédimentation et donc on les retrouve rarement dans les roches sédimentaires.
Par contre, des inclusions tels que les zircon qui résistent bien aux traitements mécaniques et chimiques seront transportés jusqu’au lieu de dépôt sédimentaire sans subir de modifications majeures.
Enfin, d’autres inclusions, surtout thorifères, de résistance moyenne ne subiront qu’une attaque chimique limitée, comme la monazite, l’apatite et le xénotime. Ces métaux lourds se déposeront en cours de transport, ou s’accumuleront dans des placers très riches.
Nous avons vu dans l’article précédent que l’uranium en milieu oxydant pouvait donné des composés complexes solubles et qui de ce fait peuvent être emportés par les eaux superficielles ou souterraines et entraînés jusqu’aux océans. Par contre, le thorium et ses isotopes sont peu solubles et ont une forte tendance à l’hydrolyse et à être précipités.
C. Classification des roches contenant des éléments radioactifs
Des observations effectuées sur des roches sédimentaires de provenances diverses ont montré que :
-
La radioactivité moyenne des sables, des grès, des calcaires et des dolomies est faible ;
-
Les argiles, qui absorbent préférentiellement les ions K+, sont fortement radioactives ;
-
La radioactivité des marnes est fonction de leur teneur en argile ;
-
Les gypses et l’anhydrite ont une très faible radioactivité.
On peut classer les roches sédimentaire sur la base du critère « radioactivité » en :
- Roches à radioactivité élevée :
-
La plupart des argiles qui constituent le support préférentiel de fixation des trois éléments radioactifs principaux, et qui peuvent, d’un autre côté, contenir des proportions importantes de matières organiques ou de phosphates, riches en uranium ;
- Les schistes noirs ;
- Les évaporites potassiques ;
- Les phosphates ;
-
Certains sables et grès riches en minéraux accessoires à uranium et thorium ;
-
Les granites potassiques et les roches qui en découlent par érosion ou métamorphisme.
- Roches à radioactivité moyenne:
- Les grès et les sables ;
- Les gneiss.
- Roches à radioactivité faible :
- Les calcaires et les dolomies ;
- Les charbons en général ;
- Les évaporites sans potassium, la halite et l’anhydrite ;
- Les roches basiques et ultrabasiques
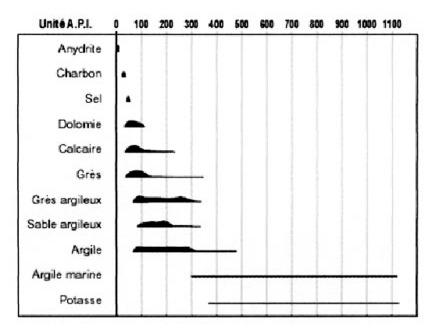
Fig. 1 – Radioactivité naturelle des principaux types de roches.
A.P.I. : unité de calibration de l’American Petroleum Institute[1]
D. Distributions des radioéléments
- Dans les calcaires
D’une manière générale, les calcaires se caractérisent par l’absence de thorium et de potassium, radioéléments liés dans les roches sédimentaires habituelles, à une phase détritique (minéraux argileux lourds). Par contre le clarke[2] de l’uranium se situe aux alentours de 2 ppm.
Ces teneurs relativement faibles s’expliquent par le fait que le calcaire provenant d’organismes aquatiques se forme dans des eaux peu profondes et bien oxygénées, milieu peu favorable à la précipitation de l’U.
Ce dernier élément se concentre dans les calcaires par des processus purement chimiques ou biochimiques. Dans ce type de roches, la radioactivité est généralement très diffuse, mais dans certains cas de calcaires impurs on peut trouver des minéraux plus ou moins actifs. Ainsi, en milieu sédimentaire carboné, les phosphates jouent souvent un rôle fondamental dans la fixation de l’uranium. En milieu marin, l’uranium est fixé par des organismes ou des carbonates d’origine biologique.
- Dans les roches carbonées
Dans les roches carbonées, on comprend les roches combustibles, d’origine organique. Elles sont constituées par les charbons et les hydrocarbures.
Rappelons que les charbons sont formés par des débris végétaux qui se sont accumulés dans l’eau par sédimentation et qui se sont transformés en milieu anaérobie, sous l’action de microbes. Les différentes phases de la transformation de ces débris végétaux en charbon sont :
-
la tourbe, qui correspond au début de leur fossilisation. Sa teneur en carbone peut atteindre 50% de son poids. Elle correspond à des dépôts récents ;
-
le lignite qui contient près de 50 à 60% de carbone. Il constitue des dépôts plus anciens ;
-
la houille dont la teneur en carbone est de 80 à 90%. Elle participe de dépôts anciens ;
-
l’anthracite contenant de 92 à 95% de carbone. C’est une roche que l’on trouve uniquement dans le Paléozoïque (Carbonifère et Permien) ;
-
le graphite correspond à la dernière augmentation de carbone (100%). Il constitue des dépôts très anciens dont les couches ont été métamorphisées.
Ces roches carbonées ont généralement des teneurs en éléments radioactifs peu significatives, du fait que les conditions favorables à leur dépôt n’existaient pas lors de leur formation.
Dans le Dakota, aux Etats-Unis, on trouve des lignites contenant 0,005 à 0,02% d’U, ce qui est très faible. Par contre, la combustion de ce lignite donne des cendres dont la teneur varie de 0,05 à 0,1%. Le lignite triasique des Vosges contient 18,8 ppm d’U (G. Jubain, 1955).
Certaines mines de lignite sont des sources de radon et de ses descendants qui peuvent être impliqués dans la genèse de cancers du poumon des travailleurs qui y sont exposés. Les taux peuvent varier fortement selon les lieux et les moments de prélèvements. Par exemple, dans trois mines de lignite de Turquie, on a relevé des concentrations variant de 50±7 à 587±16 Bq/m³ d’air.
La combustion de la houille utilisée dans les centrales thermiques émet des fumées qui sont acides et polluantes. Elles contiennent notamment des traces de vapeur de mercure et de métaux lourds et/ou radioactifs. Les cendres, résidus solides de la combustion du charbon, de ces centrales sont également chargées de métaux lourds avec des traces parfois significatives d’éléments radioactifs, atteignant de 20 à 120 ppm (U, Th, Ra…). Celles-ci accumulées sur les crassiers, sur plusieurs mètres d’épaisseur, sont exposées au vent et à la pluie. Elles sont utilisées comme fond de couche routière, matériaux de remblai ou de construction, avec le risque de polluer les nappes phréatiques.

Fig. 1 – Formation du charbon en 4 étapes
D’après compilation WEB et livres de SVT
Les hydrocarbures naturels sont des composés organiques comprenant des atomes de carbone et d’hydrogène, dont la formule brute est CnHm. Ils peuvent être :
-
des bitumes libres (gaz, pétroles, cires minérales, asphalte…) solubles dans des solvants particuliers ;
-
des bitumes potentiels (pyrobitumes, schistes bitumineux) incorporés dans certaines roches (roches mères) dont ils sont extraits par distillation.
La teneur de ces roches en U est très variable, de quelques millièmes de ppm à quelques centaines de ppm. Cet élément dont on ne sait pas sous quelle forme il s’incorpore à la roche, est soluble dans des acides et sa teneur est fonction du pourcentage de matière organique. Les schistes les plus uranifères sont noirs du fait de leur forte teneur en matière organique. Ils sont généralement riches en sulfures et pauvres en carbonates.
Certains schistes bitumineux sont parmi les roches sédimentaires les plus radioactives. Leur teneur peut varier de 0,001 à 0,035% d’U comme ceux des Etats-Unis. Les schistes bitumineux les plus anciens sont les plus radioactifs. Les plus jeunes, d’âge inférieur au Secondaire, le sont beaucoup moins.
3. Dans les marnes et argilites
Les argiles sont généralement fortement radioactives. Elles contiennent du 40K dont les nombreuses charges négatives absorbent le Th et l’U. Il est donc possible, si l’on connaît le contexte géologique, de procéder à une analyse quantitative de la teneur en argile d’une roche en établissant le dosage des trois radioéléments fixés. Par contre, le radium est mobile et se retrouve dans tous les terrains. Pour obtenir une interprétation valable, il faudra mesurer la part du radium dans la radioactivité naturelle.
Les causes de la radioactivité des argiles sont diverses :
- Il s’agit d’argiles potassiques ;
-
Les argiles ne sont pas potassiques, mais elles sont accompagnées de minéraux accessoires à potassium, uranium et thorium ;
-
A l’origine non radioactives, les argiles ont absorbé des cations comportant uranium et thorium ;
-
Certains types lithologiques sont de par leur nature radioactifs : niveaux de sels potassiques, hard-ground phosphatés, grès micacés de la mer du Nord.
Les marnes seront plus ou moins radioactives selon la proportion argile / calcaire.
4. Dans les phosphates
Certains gisements de phosphates sont des marnes ou des craies phosphatées qui se sont formés en bordure de continent ou en mer peu profonde. Le phosphate de calcium, Ca3(PO4)2, est d’origine biochimique et résulte de l’accumulation de squelettes et de cadavres d’organismes vivants. On trouve généralement de l’U diffus dans tous les phosphates et pratiquement pas de Th. Les phosphates marins sont plus riches que les phosphates continentaux. Il semble que la teneur en U augmente avec celle de la matière phosphatée, et qu’elle varie en raison inverse de la teneur en carbonate de calcium (CaCO3).
Les phosphates sédimentaires contiennent de l’U à des teneurs variables entre 50 et 200 ppm le plus souvent en équilibre avec ses descendants radioactifs, notamment le 226Ra. Comme ce dernier génère du 222Rn, les phosphates constituent une source de radon. La production d’acide phosphorique H3PO4 à partir des phosphates se fait principalement par attaque chimique à l’aide de l’acide sulfurique H2SO4. Le résidu solide de cette réaction, le phosphogypse, (CaSO4,2H2O) contient 236Ra à des activités spécifiques variables entre 300 et 900 BqKg-1. Il est également une source émanatrice de radon. Une partie du radon reste bloqué dans la structure cristalline, une autre diffuse vers l’extérieur, donnant naissance aux émanations (Berrada M. et al, voir bibliographie).
Dans le Bassin de Mons, il existe une région (Cuesmes – Mesvin – Ciply) où affleurent des craies phosphatées datées du Maestrichtien inférieur. Ces formations sont bien connues des géologues du fait de leurs intérêts économique (phosphates), stratigraphique et paléontologique (Mosasaures). Les carrières souterraines de La Malogne ont été creusées, à la fin du XIXe siècle, sous le territoire de Ciply, afin de permettre l’exploitation des craies phosphatées. Cette assisse, d’une épaisseur variant de 3 à 8 mètres, est constituée de calcarénite grise ou brune formée de nombreux grains plus ou moins phosphatés noyée dans une matrice crayeuse bioturbée, intercalée entre un hardground au sommet et le poudingue de Cuesme à la base, formé d’une accumulation de galets phosphatés et de débris fossiles. Cette craie contient en moyenne 20 à 25% de grains de phosphate de chaux, dont la teneur en U est loin d’être négligeable.
Des études sur la sismicité du Bassin de Mons y ont été menées par nos scientifiques (Charlet et al, 1990) et ont permit de classer la zone parmi les plus riches en radon de la région de Mons.
La teneur moyenne globale de l’U dans les phosphates varie dans de larges proportions selon leur origine :
| Origine | Teneur en ppm |
| Maroc | 200 à 350 |
| Tunisie | 90 à 250 |
| Algérie | 200 à 250 |
| Egypte | 100 |
| Floride | 10 à 1.500 |
| France | 20 à 150 |
-
Berrada, Boujrhal F.Z., Choukri A., Khoukhi T.EL, Iraqi M.R. – Emanation radon de phosphates sédimentaires et phosohogypses correspondants, in Radon et gaz rares dans les sciences de la Terre et de l’environnement, Actes du Colloque International sur la Géochimie des Gaz (3-6/10/1990), pp. 253-258.
-
Chapellier, Mari J.-L. – Principe de base – Cours on line de géophysique – Institut Français du Pétrole, http://www-ig.unil.ch/cours/pdf/doc_res/res_f.pdf
-
Coppens (1957) – La radioactivité des roches, PUF, « Que sais-je ?», N° 741.
-
Doremus, Kotzmann-Routier V., Quinif Y., Charlet J.-M., Flemal J.-M. – La pollution domestique par le radon – Etudes de cas : région de Mons, région de Monceau-en-Ardenne (Belgique), in Radon et gaz rares dans les sciences de la Terre et de l’environnement, Actes du Colloque International sur la Géochimie des Gaz (3-6/10/1990), pp. 33-45.
-
Pomerol , Fouet R. (1953) – Les roches sédimentaires, PUF, « Que sais-je ?», N° 595.
- http://www.solem.ch/Tunnel/didacticiel/reconnaissances/reconnaissances/MethGeophys/Radioactivitedescription.htm
[1] A.P.I. est une unité de calibration utilisée par les pétroliers dans les diagraphies de rayonnement γ. Cette unité est basé sur la radioactivité artificielle d’un bloc de béton situé à l’Université de Houston (Texas) et qui correspond à 200 A.P.I. Cette valeur a été choisie car elle correspond à deux fois la radioactivité d’un schiste typique. Les valeurs du rayonnement γ naturel sont exprimées en unité API et ensuite recalculée en cps (coups par seconde).
[2] Clarke : teneur moyenne d’un élément chimique dans la croûte terrestre, exprimée en g/t ou ppm (partie pour million).